
INTRODUCTION TO SICKLE CELL ANEMIA

- Sickle Cell Anemia falls under the broader category of sickle cell disease (SCD), which encompasses a group of inherited red blood cell disorders.
- Hemoglobin is a protein responsible for carrying oxygen in red blood cells. In Sickle Cell Anemia, a single amino acid change in the hemoglobin beta chain leads to the formation of hemoglobin S (HbS).
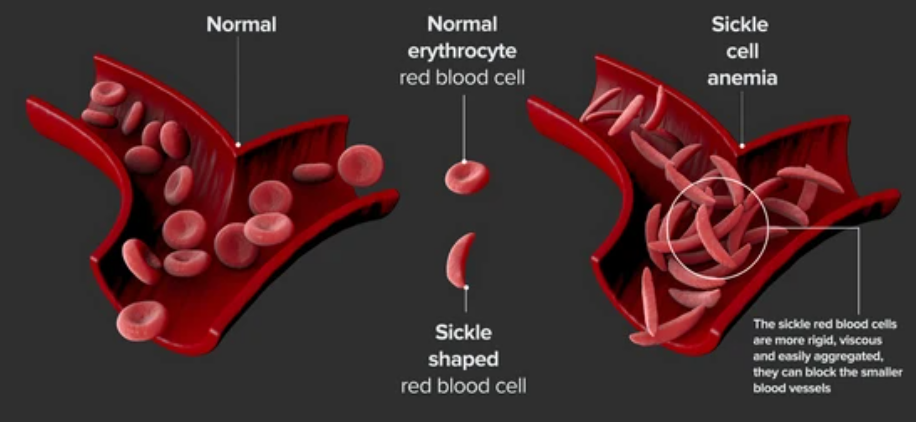
- Under certain conditions, such as low oxygen levels or dehydration, HbS molecules can aggregate and distort the shape of red blood cells, causing them to become rigid and assume a sickle shape.
- The sickle-shaped cells can get trapped in blood vessels, impeding blood flow and causing pain, tissue damage, and potentially life-threatening complications.
- Sickle Cell Anemia primarily affects individuals of African, Mediterranean, Middle Eastern, and South Asian descent, but it can occur in any population.
- The severity of the disease varies, with some individuals experiencing mild symptoms while others face frequent crises and chronic health issues.
- Common symptoms include fatigue, pain episodes (“crises”), anemia, jaundice, and increased vulnerability to infections.
GENETICS AND INHERITANCE
- Explanation of autosomal recessive inheritance : Autosomal recessive inheritance requires inheriting two mutated genes to express a trait. Sickle Cell Anemia results from a mutated HBB gene causing abnormal hemoglobin S (HbS) production. Carriers have one normal and one mutated allele, while two mutated alleles lead to the disorder in offspring.
- Role of hemoglobin in normal and sickled cells : Hemoglobin carries oxygen; normal cells have HbA. Sickle Cell Anemia’s HbS leads to rigid, obstructive cells causing pain and damage.
- Types of hemoglobin (HbA, HbS, etc.) : Hemoglobin types: HbA (normal), HbS (causing sickling), and HbF (fetal, easing symptoms). HbF remains higher in some Sickle Cell patients.
MOLECULAR BASIS OF SICKLE CELL ANEMIA
- Mutation in HBB Gene:
- The Genetic Trigger: Sickle Cell Anemia traces back to a mutation within the HBB gene, which encodes hemoglobin.
- Abnormal Hemoglobin Synthesis: This genetic alteration prompts the production of an abnormal form of hemoglobin called hemoglobin S (HbS).
B. Substitution of Amino Acids in Hemoglobin Beta Chain:
- A Tiny Swap: HbS differs from normal hemoglobin (HbA) due to a single amino acid substitution in the beta chain.
- Valine Replacing Glutamic Acid: Specifically, valine replaces glutamic acid in the beta chain, leading to altered behavior of HbS.
C. Hemoglobin Polymerization and Cell Deformation:
- Chain Formation: HbS molecules tend to aggregate and form long chains when oxygen levels drop or other stressors occur.
- Altered Cell Shape: These chain formations drive the polymerization of hemoglobin, causing red blood cells to take on a distorted, sickle-like shape.
- Impaired Blood Flow: The deformed cells’ rigidity and tendency to block blood vessels lead to reduced circulation, tissue damage, and the characteristic complications seen in Sickle Cell Anemia.
SYMPTOMS OF SICKLE CELL ANEMIA
- Painful Crises: Recurrent episodes of pain occur due to blocked blood vessels.
- Anemia: Fatigue and weakness result from a shortage of healthy red blood cells.
- Jaundice: Yellowing of the skin and eyes due to increased breakdown of sickled cells.
- Infections: Higher susceptibility to infections due to compromised immune function.
- Growth and Development: Delayed growth and development can affect children.
- Variable Severity: Symptoms range from mild to severe among individuals.
- Organ Damage: Prolonged disease can lead to organ damage and complications.
- Effective Management: Treatment aims to alleviate symptoms and prevent complications.
DIAGNOSIS AND SCREENING
- Hemoglobin Electrophoresis:
- Detecting Hemoglobin Variants: Hemoglobin electrophoresis is a diagnostic technique that separates different types of hemoglobin based on their electrical charge.
- Identifying Abnormal Hemoglobins: This method helps distinguish between various hemoglobin variants, including HbS, enabling the diagnosis of Sickle Cell Anemia and other hemoglobinopathies.
- Genetic Testing:
- Unveiling Genetic Makeup: Genetic testing involves analyzing an individual’s DNA to identify specific mutations associated with Sickle Cell Anemia.
- Carrier Identification and Prenatal Diagnosis: Genetic testing not only confirms the presence of the HbS gene but also aids in identifying carriers and facilitating prenatal diagnosis.
Newborn Screening Programs:
- Early Detection: Many regions have implemented newborn screening programs that include testing for Sickle Cell Anemia. This enables early identification and intervention in affected infants.
- Timely Management: Early diagnosis through newborn screening allows for prompt medical management, reducing the risk of complications and ensuring better outcomes for affected individuals.
MANAGEMENT AND TREATMENT:

A. Symptomatic Treatment:
- Pain Relief: Managing Sickle Cell Anemia often involves pain management through medications to alleviate pain during crises.
- Hydration: Staying well-hydrated helps prevent sickling of red blood cells and reduces the likelihood of painful episodes.
B. Blood Transfusions and Iron Overload:
- Blood Transfusions: Periodic transfusions can provide healthy red blood cells, improving oxygen delivery and reducing complications.
- Iron Overload Management: Frequent transfusions can lead to excess iron accumulation, requiring iron chelation therapy to prevent organ damage.
C. Hematopoietic Stem Cell Transplantation:
- Curative Approach: Hematopoietic stem cell transplantation involves replacing the bone marrow with healthy donor cells, potentially curing Sickle Cell Anemia.
- Challenges and Considerations: This approach is complex and may be limited by donor availability and compatibility.
D. Emerging Therapies:
- Gene Therapy: Researchers are exploring gene therapy to replace the mutated HBB gene with a functional version, aiming for a lasting cure.
- Targeted Drugs: New drugs targeting specific mechanisms involved in sickling are under investigation, offering potential treatment options.
CURRENT RESEARCH AND FUTURE DIRECTIONS
A. Advancements in Understanding the Disease:
- Genetic Insights: Ongoing research continues to uncover deeper genetic mechanisms behind Sickle Cell Anemia, aiding in more precise diagnosis and treatment.
- Disease Pathways: Scientists are unraveling the intricate pathways that lead to cell sickling, offering potential targets for new therapies.
B. Potential Curative Treatments:
- Gene Editing: CRISPR-based gene editing techniques hold promise for correcting the underlying genetic mutation responsible for Sickle Cell Anemia.
- Innovative Therapies: Advancements in stem cell research and gene therapy aim to develop curative approaches that could eliminate symptoms.
C. Public Health Strategies and Awareness:
- Newborn Screening: Expanding newborn screening programs globally can ensure early diagnosis and timely management.
- Education and Advocacy: Raising awareness about Sickle Cell Anemia helps reduce stigma, improve patient outcomes, and promote research funding.
CONCLUSION
In summary, Sickle Cell Anemia, resulting from a genetic mutation in the HBB gene, exemplifies the complex relationship between genetics and health. Amino acid substitution in hemoglobin’s beta chain triggers abnormal polymerization, distorting red blood cells. Ongoing research offers potential through gene editing and advanced therapies. Public health measures, including newborn screening and awareness campaigns, play a vital role. As these efforts converge, a future with improved treatments and, possibly, a cure for Sickle Cell Anemia comes into focus.
Share this:

Discover more from ZOOLOGYTALKS
Subscribe to get the latest posts sent to your email.